IN A NUTSHELL
1. About This Work
Over the last decade, incredible amounts of biological sequences have been accumulated, benefitting from the swift advances in high-throughput sequencing technologies. Currently, many efforts are being made to regularly and rapidly generate more and more biological sequences. For example, the 100,000 Genomes Project by National Health Service (NHS) in UK aims at sequencing 100,000 genomes from around 70,000 people. These participants include patients with rare disease, plus their families, and patients with cancer. Along with the flourishing growth of these datasets, machine learning has been more and more widely applied to gain insights into biological systems and complex diseases, thus becoming integral to current biological research. Across various such applications, a common and vital step is to transform original biological sequences into machine learning operable features with exposure of their intrinsic statistical relationships, based on which predictive patterns could be further recognized. Obviously, feature extraction procedure is significantly important and even more complicated to machine learning studies in biology, due to the existing gap between the biological sequences and machine learning operable vectors/matrix.
To bridge this gap, many computational efforts emerge with the purpose of formulating biological sequences into discrete or numerical vectors that could capture sequences’ intrinsic patterns and characteristics. These previous works summarize different types of biological sequence based extraction algorithms, accordingly implement them, and finally provide web-based or/and standalone toolkits to expedite generation of such features. While each of those toolkits has its own distinct merits, they usually share some limitations, summarized in threefold aspects: 1) lack of computing power in web servers for feature generating which is reflected by the limited number of sequences per submission; 2) lack of extensibility in web servers to customize features, some of which even don’t support numerical parameter adjustment online; and 3) deficiency in integration of heterogeneous feature to provide all-in-one service.
To overcome the above shortcoming, we design and implement a distributed framework, termed DIFFUSER, for efficiently and comprehensively generate a broad spectrum of heterogeneous features based on biological sequences. The contribution of DIFFUSER could be summarized into threefold aspect as follows. First, DIFFUSER employs a brand-new and self-developed distributed architecture to take advantages of a cluster of computing nodes, instead of using a single server, to largely improve the throughput of feature generation. Compared to single server based applications, DIFFUSER speeds up the entire feature generating procedure by n times, where n represents the numbers of computing nodes in the cluster. Benefitting from the framework's extensibility, DIFFUSER's computing power could be easily extended in linear scale, by adding in more computing facilities with simple configuration. Second, DIFFUSER represents the most comprehensive feature generator, which covers the largest number of features in a broadest spectrum to provide all-in-one service. Last, DIFFUSER provides a user-friendly web-based server and a standalone toolkit, both of which provide the same functions to generate all types of features with full support of feature customization.
ARCHITECTURE
1. Architecture of the distributed web server
Despite easiness for non-technical researchers, web-based server also brings some challenges on the server's computing capability, due to the fact the web-based server process all the requests using its own central computing power. This situation becomes more severe when the jobs (such as PSSM-based feature generation) are time consuming or job requests breakthrough within a short period. To avoid such dilemma, current existing feature generating servers set a maximal number (usually less than 500) of sequences per submission, which eases the servers burden to some extent, but at the same time lowers the overall user experience and limits their applications in high throughput manner.
Considering of the intrinsic requirement of large datasets in machine learning application, especially under current background of big data, it's vitally important and extremely necessary to construct new-generation web servers with the power of processing large-scale data. To enable the DIFFUSER server to process biological sequences in genome-scale, we design and implement a distributed framework in virtue of distributed/parallel computing and distributed file storage.
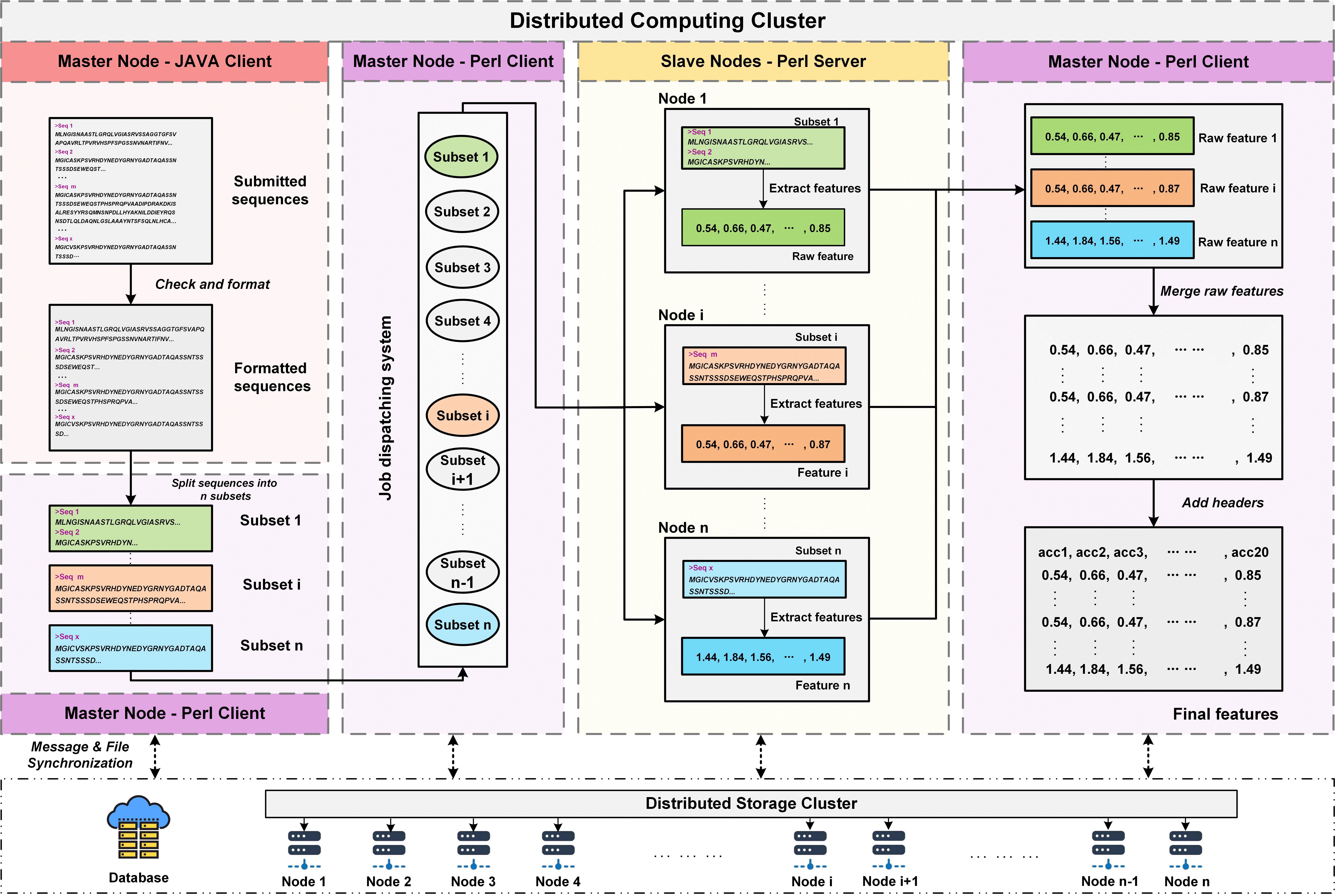
The distributed computing cluster is responsible for executing the feature generating task in parallel, by dividing the feature generating task into many sub-tasks. Each of the sub-tasks will be executed by a single computer node, and the results of these sub-tasks will be merged into a final feature file. Specifically, the distributed computing cluster consists of two types of nodes: a master node and multiple slave nodes. Deployed in the master node, web-based server provides users a graphical and user-friendly interface to submit their target biological sequences after selecting some or all of features and specifying according parameters. Once the sequences are submitted, they will be passed to the backend of the web-based server (developed by JAVA web developing suite) for further check and format. The legal submitted sequences in a uniformed form will be then forwarded to a job dispatching system, which is developed based on Gearman using Perl language and deployed across the master node and the slave nodes. The submitted sequences will be split into subsets and then put into the job queue by the master node. The slave nodes with idle threads will proactively fetch jobs from the job queue, and accordingly execute feature generating job separately for the individual subsets. Once a slave node finishes the feature generating procedure for a subset, it will notify the client of the job dispatching system in the master node. After all the subsets of a whole submitted sequence job are finished, the client of the job dispatching system in the master node will be responsible to merge all of these sub-feature sets together into a final feature file.
The distributed file storage is responsible for sharing temporary and final files within the distributed computing cluster. Developed based on FastDFS, it has been deployed in all the distributed computing cluster nodes. Each node could directly drop its files during the feature generating procedure, and those files will be automatically duplicated and shared by other nodes. Additionally, a database, developed based on MySQL, is used to record and share status of sub-jobs. In this way, the nodes within the distributed computing cluster could run in parallel but cooperate well with each other as an overall distributed system.
2. Architecture of the Toolkit
Besides of web-based server, the DIFFUSER standalone toolkit is provided to enable users to customize large-scale features on their own computing facility. This would be indispensable if feature extraction procedure need to be automatically executed or included into the sequence analysis pipeline. Previous existing toolkits are developed by various programming languages, such as Pse-in-One in Python, protr in R, POSSUM in Perl and Python, and PseAAC-General in C/C++, which sets an obstacle in front of users if they need to generate a group of features across different features. The situation becomes even worse, where users are required to write program language-specific scripts , such as Python fundamental needed in use of propy and R fundamental in protr. To fill this gap, the DIFFUSER standalone toolkit covers as many types of features as possible in accordance with its web server, with full support of self-customization on feature generating. As a pure Python developed toolkit, it is easy to configure (only several Python libraries are required and could be easily installed using several commands) and simple to use on various operating systems, such as executed on Unix/Linux, Windows and Mac OS. To generate a feature for a given dataset, only a line of Shell command is needed, which could also be directly obtained from the web server result page, if users have tried an example online.
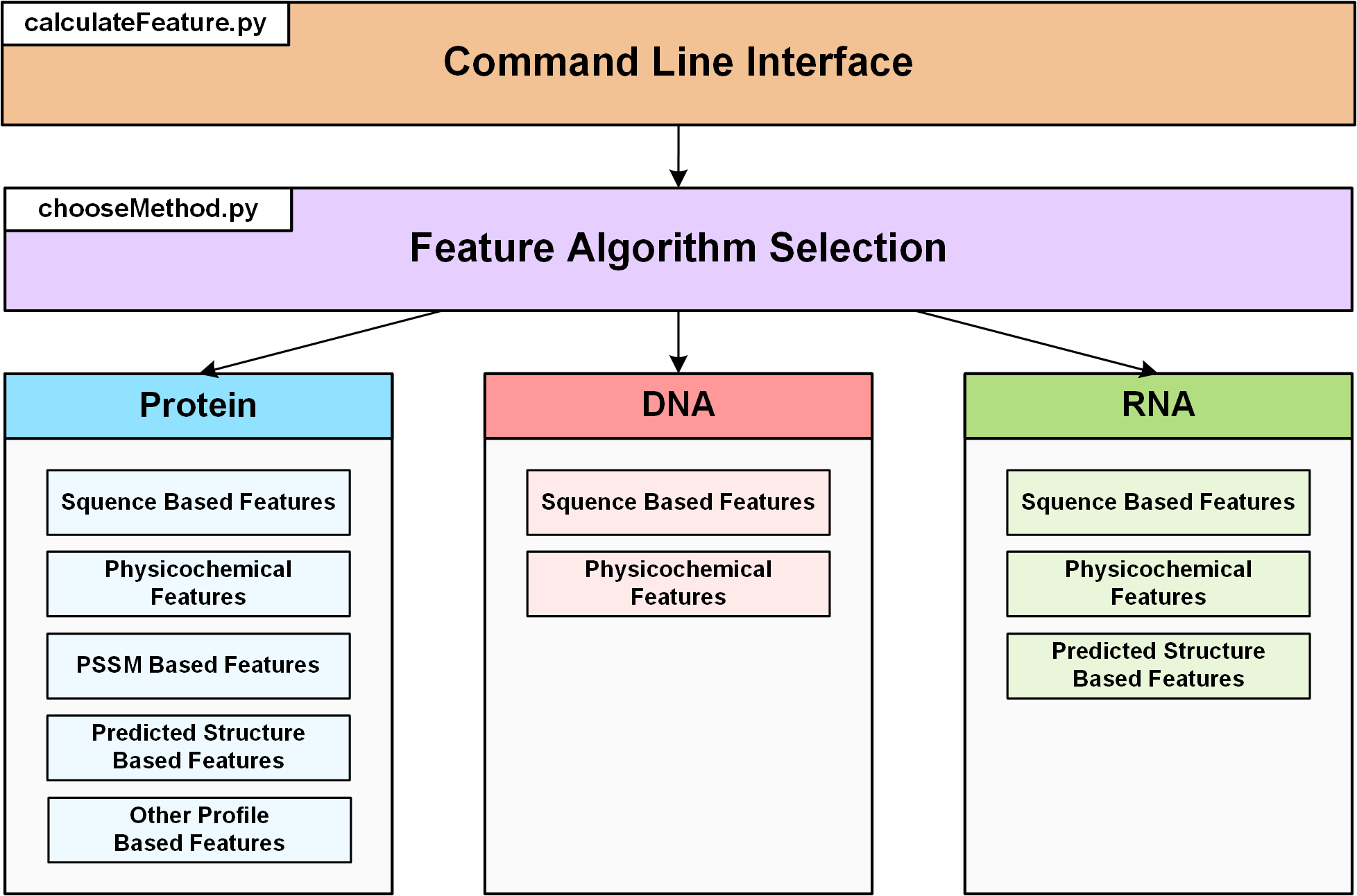
We modified, integrated and refactored the source code of POSSUM, iFeature, Pse-in-One and BioSeq-Analysis to construct the DIFFUSER standalone toolkit. The major components of the toolkit are briefly described as follows:
-
Command Line Interface: This module is made available to provide a universal and user-friendly command line interface, via which users can effectively interact with the toolkit. This module allows users to specify and apply different parameters and it invokes the descriptor generating process. -
Feature Algorithm Selection: This module can be used to select various algorithm for generating wide-ranging features, including Protein descriptors, DNA descriptors and RNA descriptors. -
Protein: This module can be used to generate various features for protein sequences. A total of five groups are Sequence Based Features, Physicochemical Features, PSSM Based features, Predicted Structure Based Features and Other Profile Based Features. -
DNA: This module can be used to generate various features for DNA sequences. A total of two groups are Sequence Based Features and Physicochemical Features. -
RNA: This module can be used to generate various features for RNA sequences. A total of three groups are Sequence Based Features, Physicochemical Features and Predicted Structure Based Features.
ONLINE WEB SERVER
1. Input
1.1 Input formats
Two types of input are allowed for DIFFUSER: sequences in FASTA format (recommended) or raw sequences.
For sequences in FASTA format, you can input as follows:
Also, the following input (which is the original formats downloaded from Uniprot database)
will be formated (without line break inside the sequence) as:
For the raw sequences, you can input as follows:
which will be formated by POSSUM as follows:
1.2 Input limits
- The length of each submitted sequence should be in the range of 9 to 5000 characters.
- Since PSSM-based feature need longer sequences to generate PSSM, the length of each submitted sequence should be in the range of 50 to 5000 characters when you choose extract PSSM-based feature.
- The DIFFUSER is a server based on the distributed framework, so the number of each submitted sequence is up to 5000.
- The submitted sequences should not contain illegal characters, such as "B", "J", "O", "U", "X" and "Z".
2 Upload a file in fasta format
Instead of inputing sequences directly in the textarea, users can also upload a file in fasta format.
Note: please don't submit sequences by textarea and file uploading simultaneously. Users should choose one or the other for each submission.
3 Select algorithms to generate descriptors
The main function of DIFFUSER toolkit is to provide an all-in-one service to generate customizable and heterogeneous features based on various biological sequences, including protein, DNA and RNA sequences. Towards this purpose, we integrate and implement XX types of features in 5 groups, which to the utmost covers the existing feature extraction algorithms designed from various aspects. Group1 represents sequence based features; Group2 represents physicochemical features; Group3 represents PSSM based features; Group4 represents predicted structure based features; Group5 represents other profile based features. Among them, there are 82 feature encodings based on Protein sequences, 20 feature encodings based on DNA sequences and 14 feature encodings based on RNA sequences.
Before selecting algorithms to generate descriptors, please note the following tips:
- " * " indicates the parameter is required.
- " * " indicates submitted sequences should be the same length.
- " * " indicates the length of sequences should larger than 50 based on PSSM-features.
4 Cache files
Some intermediate files (.pssm files, .dis files, .ss2 files and .spXout files) are used when PSSM-based features and predicted structure composition derived features are calculated. Since generating these intermediate files is very time consuming, the DIFFUSER server caches these generated intermediate files. When the user submitted the sequence which is same as the former submitted sequences, the corresponding intermediate file do not need to be regenerated, just take them from the cache file to further improve computing efficiency.
Specially, the intermediate file corresponding to the example fasta file is cached. Therefore, example fasta is faster than normal when generating features.
5 Output
There exist 3 types of output pages for the result of computation: result page, warning page and error page.
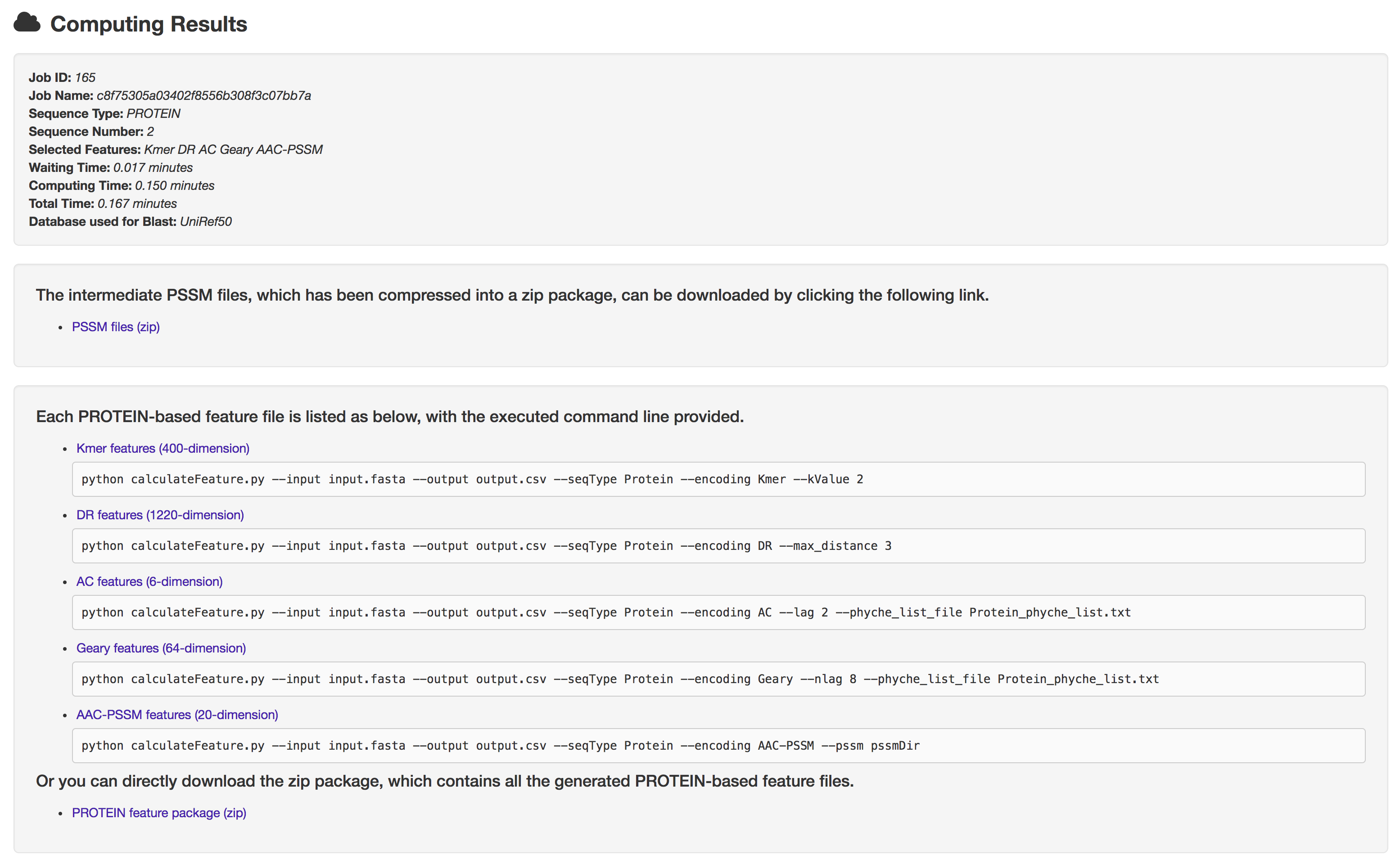
5.1 Result page
The result page consists of 3 parts: job information, intermediate files download and feature files download.
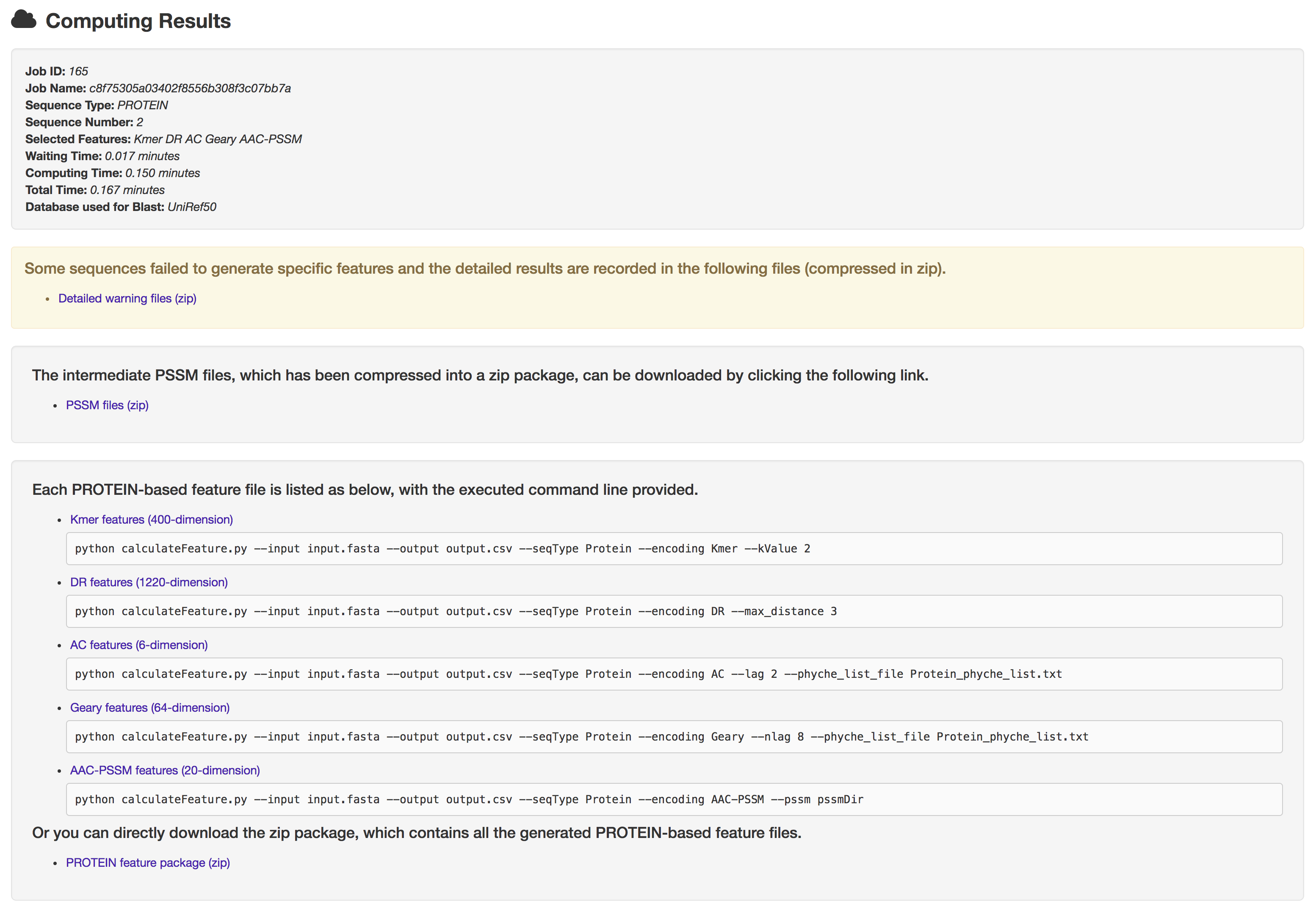
5.2 Warning page
Should any sequences be failed to generate specific features (this probability is extremely small, but is finite.). Users will receive a warning page, which contains 4 parts: job information, detailed warning files, intermediate files of the rest sequences and feature files of the rest sequences.
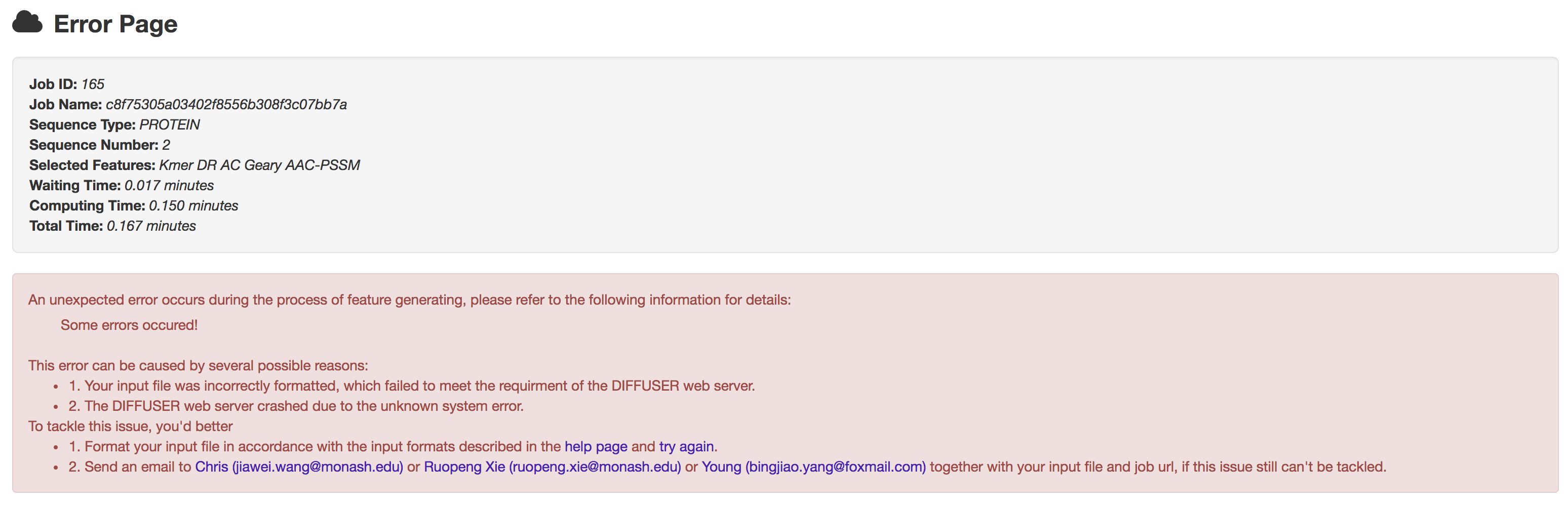
5.3 Error page
Should the computing process stop abnormally due to any unexpected system errors, users will get an error page which contains 2 parts: job information and error details.
STANDALONE SOFTWARE
1. Overview
The source codes of DIFFUSER can be downloaded at the DOWNLOAD page.
2. Using DIFFUSER
For users who prefer to apply their own parameter settings for specific research purposes and users who have the capacity to perform high throughput generation of intermediate files (such as .pssm files, .dis files, .ss2 files, .spXout files and so on) for a very large dataset using their local computers, an open source standalone software toolkit is also available. The standalone version of DIFFUSER was purely developed using Python, and can be executed on Unix/Linux, Windows and Mac OS. As an open source software, users can access, modify and redistribute the source codes, allowing users to tailor DIFFUSER according to their specific requirements.
2.1 System Requirements
-
Operating systems:
Windows,Unix/Linux,Mac OS - Dependencies:
2.2 File Description in the download directory
-
Protein: The folder contains the source code, example file and required extra data for generating protein features.*.py: The python script files used to generate protein descriptors.data: The data folder, which contains example input file used to generate descriptors.example: The example folder, which contains example input file used to generate descriptors.pssm_files: The folder where PSSM files are stored.disorder_files: The folder where .dis files are stored.ss_files: The folder where .ss2 files are stored.spX_files: The folder where .spXout files are stored.pdt: The pdt folder, which used to generate the intermediate file required for PDT features.
-
DNA: The folder contains the source code, example file and required extra data for generating DNA features.*.py: The python script files used to generate DNA descriptors.data: The data folder, which contains example input file used to generate descriptors.example: The example folder, which contains example input file used to generate descriptors.
-
RNA: The folder contains the source code, example file and required extra data for generating RNA features.*.py: The python script files used to generate RNA descriptors.data: The data folder, which contains example input file used to generate descriptors.example: The example folder, which contains example input file used to generate descriptors.
-
utils: The folder contains three scripts to generate intermediate files. -
docs: The folder used to store help documents.userguide.pdf: The detailed description file for DIFFUSER standalone toolkit.
-
calculateFeature.py: A python script facilating users to invoke and run DIFFUSER standalone toolkit. -
checkFile.py: A python script used to check input files whether have illegal characters. -
chooseMethod.py: A python script used to invoke feature generation file in Protein, DNA and RNA folder. -
readFile.py: A python script used to read input files, intermediate files and so on.
2.3 Usage
Protein:
For Kmer algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding Kmer --kValue 2
-
Tips:Parameter kValue in Kmer, kValue must be an integer and the range of kValue is preferably between 1 and 6. -
References (PubMed ID):19046430 29272359
For DR algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding DR --max_distance 3
-
Tips:Parameter max_distance in DR, max_distance must be an integer and the range of max_distance is preferably between 1 and 10. -
References (PubMed ID):24564580 29272359
For DP algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding DP --max_distance 3 --cp cp_14
-
Tips:Parameter max_distance in DP, max_distance must be an integer and the range of max_distance is preferably between 1 and 10. The parameter cp is the reduced alphabet approach to significantly cut down the dimension of the PseAAC vector. Choose one of the four: "cp_13, cp_14, cp_19, cp_20". -
References (PubMed ID):25184541 29272359
For EAAC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding EAAC --sliding_window 5
-
Tips:Parameter sliding_window in EAAC, sliding_window must be an integer and smaller than minimum length of submitted sequences. -
References (PubMed ID):29528364
For CKSAAP algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding CKSAAP --gap 5
-
Tips:Parameter gap in CKSAAP, gap must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 2. -
References (PubMed ID):17316561 29528364
For DDE algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding DDE
-
References (PubMed ID):26406767 29528364
For GAAC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding GAAC
-
References (PubMed ID):21551145 29528364
For EGAAC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding EGAAC --sliding_window 5
-
Tips:Parameter sliding_window in EGAAC, sliding_window must be an integer. -
References (PubMed ID):29528364
For CKSAAGP algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding CKSAAGP --gap 5
-
Tips:Parameter gap in CKSAAGP, gap must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 2. -
References (PubMed ID):29528364
For GDPC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding GDPC
-
References (PubMed ID):29528364
For GTPC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding GTPC
-
References (PubMed ID):29528364
For BINARY algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding BINARY
-
References (PubMed ID):21829559 30351377
For NUM algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding NUM
-
References (PubMed ID):30351377
For AC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding AC --phyche_list_file Protein/example/protein_phyche_list.txt --extra_index_file Protein/example/protein_indices.txt --lag 2
-
Tips:Parameter lag in AC, lag must be an integer and greater than 0 and smaller than minimum length of submitted sequences. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):19706744 29272359
For CC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding CC --phyche_list_file Protein/example/protein_phyche_list.txt --extra_index_file Protein/example/protein_indices.txt --lag 2
-
Tips:Parameter lag in CC, lag must be an integer and greater than 0 and smaller than minimum length of submitted sequences. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):19706744 29272359
For ACC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding ACC --phyche_list_file Protein/example/protein_phyche_list.txt --extra_index_file Protein/example/protein_indices.txt --lag 2
-
Tips:Parameter lag in ACC, lag must be an integer and greater than 0 and smaller than minimum length of submitted sequences. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):19706744 29272359
For PDT algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding PDT --lamada 1
-
Tips:Parameter lamada in PDT, lamada must be an integer and the range of lamada is preferably between 1 and 15. -
References (PubMed ID):23029559 29272359
For Moran algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding Moran --phyche_list_file Protein/example/protein_phyche_list_auto.txt --nlag 8
-
Tips:Parameter nlag in Moran, nlag must be an integer and greater than 0 and and greater than 0 and smaller than that minimum length of submitted sequences minus 1. The format of parameter phyche_list_file should be consistent with the corresponding sample file. -
References (PubMed ID):11043931 29528364
For Geary algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding Geary --phyche_list_file Protein/example/protein_phyche_list_auto.txt --nlag 8
-
Tips:Parameter nlag in Geary, nlag must be an integer and greater than 0 and and greater than 0 and smaller than that minimum length of submitted sequences minus 1. The format of parameter phyche_list_file should be consistent with the corresponding sample file. -
References (PubMed ID):16261547 29528364
For NMBroto algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding NMBroto --phyche_list_file Protein/example/protein_phyche_list_auto.txt --nlag 8
-
Tips:Parameter nlag in NMBroto, nlag must be an integer and greater than 0 and and greater than 0 and smaller than that minimum length of submitted sequences minus 1. The format of parameter phyche_list_file should be consistent with the corresponding sample file. -
References (PubMed ID):3359010 29528364
For PAAC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding PAAC --phyche_list_file Protein/example/protein_phyche_list.txt --extra_index_file Protein/example/protein_indices.txt --lamada 2 --weight 0.1
-
Tips:Parameter lamada in PAAC, lamada must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 1. The parameter weight must be a number and greater than 0. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):23426256 29528364
For APAAC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding APAAC --phyche_list_file Protein/example/protein_phyche_list.txt --extra_index_file Protein/example/protein_indices.txt --lamada 2 --weight 0.1
-
Tips:Parameter lamada in APAAC, lamada must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 1. The parameter weight must be a number and greater than 0. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):23426256 29528364
For CTDC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding CTDC
-
References (PubMed ID):7568000 29528364
For CTDT algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding CTDT
-
References (PubMed ID):7568000 29528364
For CTDD algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding CTDD
-
References (PubMed ID):7568000 29528364
For CTriad algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding CTriad
-
References (PubMed ID):17360525 29528364
For KSCTriad algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding KSCTriad --kValue 2
-
Tips:Parameter kValue in KSCTriad, kValue must be an integer and 2kValue+3 should smaller than minimum length of submitted sequence. -
References (PubMed ID):17360525 29528364
For SOCNumber algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding SOCNumber --nlag 8
-
Tips:Parameter nlag in SOCNumber, nlag must be an integer and smaller than that minimum length of submitted sequence minus 1. -
References (PubMed ID):11097861 29528364
For QSOrder algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding QSOrder --nlag 8 --weight 0.1
-
Tips:Parameter nlag in QSOrder, nlag must be an integer and smaller than that minimum length of submitted sequence minus 1. The parameter weight must be a number and greater than 0. -
References (PubMed ID):11097861 29528364
For KNNprotein algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding KNNprotein --train Protein/example/example_train_protein.fasta --label Protein/example/label.txt
-
Tips:Parameter train and label are required in KNNprotein and the formats of parameter train and label should be consistent with the corresponding sample file. -
References (PubMed ID):29528364
For KNNpeptide algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding KNNpeptide --train Protein/example/example_train_peptide.fasta --label Protein/example/label.txt
-
Tips:Parameter train and label are required in KNNpeptide and the formats of parameter train and label should be consistent with the corresponding sample file. -
References (PubMed ID):11097861 29528364
For AAINDEX algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding AAINDEX
-
References (PubMed ID):18625080 29528364
For BLOSUM62 algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding BLOSUM62
-
References (PubMed ID):21408064 29528364
For ZSCALE algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding ZSCALE
-
References (PubMed ID):22720073 29528364
For EBGW algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding EBGW --lamada 8
-
Tips:Parameter lamada in EBGW, lamada must be an integer and greater than 0 and smaller than minimum length of submitted sequences. -
References (PubMed ID):19706744 29272359
For PseKRAAC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding PseKRAAC --kValue 2 --subtype lambda-correlation --type type1 --gap_lambda 1 --raactype 2
-
Tips:Parameter kValue (i.e. 1, 2 and 3) are available. Parameter subtype is the feature types for protein sequences analysis, choose one of two: "g-gap, lambda-correlation". Parameter gap_lamabda is the gap value or lambda value for the "g-gap" model or "lambda-correlation" model in PseKRAAC, the range of gap_lamabda is between 1 and 10. Parameter type need choose one from "type1", "type2", "type3A", "type3B", "type4", "type5", "type6A", "type6B", "type6C","type7", "type8", "type9", "type10", "type11", "type12", "type13", "type14", "type15", "type16". The value of corresponding parameter raactype can refer to Protein/index_list.py. -
References (PubMed ID):27565583 29528364
For AAC-PSSM algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding AAC-PSSM --pssm Protein/example/pssm_files/
-
Tips:Parameter pssm in AAC-PSSM, pssm denotes the folder of pssm files. -
References (PubMed ID):20600567 28903538
For D-FPSSM algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding D-FPSSM --pssm Protein/example/pssm_files/
-
Tips:Parameter pssm in D-FPSSM, pssm denotes the folder of pssm files. -
References (PubMed ID):23747746 28903538
For smoothed-PSSM algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding smoothed-PSSM --pssm Protein/example/pssm_files/ --smoothing_window 7 --sliding_window 50
-
Tips:Parameter pssm in smoothed-PSSM, pssm denotes the folder of pssm files. Parameter smoothing_window denotes the size of smoothing window and should be an odd number. Parameter sliding_window denotes the size of sliding window and must be larger than maximum value of smoothing_window and smaller than minimum length of submitted sequence. -
References (PubMed ID):19091029 29186295
For AB-PSSM algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding AB-PSSM --pssm Protein/example/pssm_files/
-
Tips:Parameter pssm in AB-PSSM, pssm denotes the folder of pssm files. -
References (PubMed ID):20855926 28903538
For PSSM-composition algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding PSSM-composition --pssm Protein/example/pssm_files/
-
Tips:Parameter pssm in PSSM-composition, pssm denotes the folder of pssm files. -
References (PubMed ID):24064423 29186295
For RPM-PSSM algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding RPM-PSSM --pssm Protein/example/pssm_files/
-
Tips:Parameter pssm in RPM-PSSM, pssm denotes the folder of pssm files. -
References (PubMed ID):20855926 28903538
For S-FPSSM algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding S-FPSSM --pssm Protein/example/pssm_files/
-
Tips:Parameter pssm in S-FPSSM, pssm denotes the folder of pssm files. -
References (PubMed ID):23747746 29547915
For DPC-PSSM algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding DPC-PSSM --pssm Protein/example/pssm_files/
-
Tips:Parameter pssm in DPC-PSSM, pssm denotes the folder of pssm files. -
References (PubMed ID):20600567 29547915
For k-separated-bigrams-PSSM algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding k-separated-bigrams-PSSM --pssm Protein/example/pssm_files/ --kValue 1
-
Tips:Parameter pssm in k-separated-bigrams-PSSM, pssm denotes the folder of pssm files. Parameter kValue must be an integer and the range of kValue is preferably between 1 and 3. -
References (PubMed ID):28903538
For tri-gram-PSSM algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding tri-gram-PSSM --pssm Protein/example/pssm_files/
-
Tips:Parameter pssm in tri-gram-PSSM, pssm denotes the folder of pssm files. -
References (PubMed ID):24594513 28903538
For EEDP algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding EEDP --pssm Protein/example/pssm_files/
-
Tips:Parameter pssm in EEDP, pssm denotes the folder of pssm files. -
References (PubMed ID):24735902 28903538
For TPC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding TPC --pssm Protein/example/pssm_files/
-
Tips:Parameter pssm in TPC, pssm denotes the folder of pssm files. -
References (PubMed ID):22545994 28903538
For EDP algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding EDP --pssm Protein/example/pssm_files/
-
Tips:Parameter pssm in EDP, pssm denotes the folder of pssm files. -
References (PubMed ID):24735902 28903538
For RPSSM algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding RPSSM --pssm Protein/example/pssm_files/
-
Tips:Parameter pssm in RPSSM, pssm denotes the folder of pssm files. -
References (PubMed ID):24067326 28903538
For Pse-PSSM algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding Pse-PSSM --pssm Protein/example/pssm_files/ --Xi 1
-
Tips:Parameter pssm in Pse-PSSM, pssm denotes the folder of pssm files. Parameter Xi must be an integer and the range of Xi is preferably between 1 and 5. -
References (PubMed ID):17586467 29547915
For DP-PSSM algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding DP-PSSM --pssm Protein/example/pssm_files/ --Alpha 5
-
Tips:Parameter pssm in DP-PSSM, pssm denotes the folder of pssm files. Parameter Alpha must be an integer and the range of Alpha is preferably between 1 and 10. -
References (PubMed ID):28903538
For PSSM-AC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding PSSM-AC --pssm Protein/example/pssm_files/ --lag 10
-
Tips:Parameter pssm in PSSM-AC, pssm denotes the folder of pssm files. Parameter lag must be an integer and the range of lag is preferably between 1 and 20. -
References (PubMed ID):19706744 29186295
For PSSM-CC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding PSSM-CC --pssm Protein/example/pssm_files/ --lag 10
-
Tips:Parameter pssm in PSSM-CC, pssm denotes the folder of pssm files. Parameter lag must be an integer and the range of lag is preferably between 1 and 20. -
References (PubMed ID):19706744 28903538
For AADP-PSSM algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding AADP-PSSM --pssm Protein/example/pssm_files/
-
Tips:Parameter pssm in AADP-PSSM, pssm denotes the folder of pssm files. -
References (PubMed ID):20600567 28903538
For AATP algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding AATP --pssm Protein/example/pssm_files/
-
Tips:Parameter pssm in AATP, pssm denotes the folder of pssm files. -
References (PubMed ID):22545994 28903538
For MEDP algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_protein.fasta --output output.csv --seqType Protein --encoding MEDP --pssm Protein/example/pssm_files/
-
Tips:Parameter pssm in MEDP, pssm denotes the folder of pssm files. -
References (PubMed ID):24735902 28903538
For Disorder algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding Disorder --path Protein/example/disorder_files/
-
Tips:Parameter path in Disorder, path denotes the folder of disorder files. -
References (PubMed ID):21267749 29528364
For DisorderB algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding DisorderB --path Protein/example/disorder_files/
-
Tips:Parameter path in DisorderB, path denotes the folder of disorder files. -
References (PubMed ID):29528364
For DisorderC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding DisorderC --path Protein/example/disorder_files/
-
Tips:Parameter path in DisorderC, path denotes the folder of disorder files. -
References (PubMed ID):29528364
For SSEC algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding SSEC --path Protein/example/ss_files/
-
Tips:Parameter path in SSEC, path denotes the folder of disorder files. -
References (PubMed ID):29528364
For SSEB algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding SSEB --path Protein/example/ss_files/
-
Tips:Parameter path in SSEB, path denotes the folder of disorder files. -
References (PubMed ID):29528364
For ASA algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding ASA --path Protein/example/spX_files/
-
Tips:Parameter path in ASA, path denotes the folder of disorder files. -
References (PubMed ID):29528364
For TA algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding TA --path Protein/example/spX_files/
-
Tips:Parameter path in TA, path denotes the folder of disorder files. -
References (PubMed ID):29528364
For LOGO algorithm:
-
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding LOGO --frequency_file Protein/example/example_peptide_frequency.txt
-
Tips:Parameter frequency_file in LOGO, frequency_file is generated by Two Sample Logo with p-value smaller than 1 and txt output format. -
References (PubMed ID):30351377
For LOGO-P-Value algorithm:
-
Description:The core idea of this algorithm is P-Value integrated into LOGO. P-Value is the probability of the difference between samples due to sampling error. In this method, we multiply that 1 minus P-Value by the value of the LOGO to generate features. -
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding LOGO-P-Value --frequency_file Protein/example/example_peptide_frequency.txt
-
Tips:Parameter frequency_file in LOGO-P-Value, frequency_file is generated by Two Sample Logo with p-value smaller than 1 and txt output format. -
References (PubMed ID):This work.
For LOGO-BLOSUM62 algorithm:
-
Description:The core idea of this algorithm is BLOSUM62 integrated into LOGO. In this method, we sum the product of the LOGO value and the similarity of each amino acid by BLOSUM62 to generate features. -
Command line:python calculateFeature.py --input Protein/example/example_peptide.fasta --output output.csv --seqType Protein --encoding LOGO-BLOSUM62 --frequency_file Protein/example/example_peptide_frequency.txt
-
Tips:Parameter frequency_file in LOGO-BLOSUM62, frequency_file is generated by Two Sample Logo with p-value smaller than 1 and txt output format. -
References (PubMed ID):This work.
DNA:
For Kmer algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding Kmer --kValue 2
-
Tips:Parameter kValue in Kmer, kValue must be an integer and the range of kValue is preferably between 1 and 6. -
References (PubMed ID):15961476 26362104
For RevKmer algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding RevKmer --kValue 2
-
Tips:Parameter kValue in RevKmer, kValue must be an integer and the range of kValue is preferably between 1 and 6. -
References (PubMed ID):15961476 18725940
For IDKmer algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding IDKmer --kValue 4 --positiveSource DNA/example/example_dna_ps.fasta --negativeSource DNA/example/example_dna_ns.fasta
-
Tips:Parameter kValue in IDKmer, kValue must be an integer and the range of kValue is preferably between 1 and 6. The positiveSource and negativeSource parameters are required. -
References (PubMed ID):14576308 20097656
For Mismatch algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding Mismatch --kValue 3 --mismatch 1
-
Tips:Parameter kValue in Mismatch, kValue must be an integer and the range of kValue is preferably between 1 and 6. Parameter mismatch in Mismatch, mismatch must be an integer and greater than 0 and smaller than kValue. -
References (PubMed ID):14990442 19642274
For Subsequence algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding Subsequence --kValue 3 --delta 1
-
Tips:Parameter kValue in Subsequence, kValue must be an integer and the range of kValue is preferably between 1 and 3. Parameter delta in Subsequence, delta must be a number and the range of delta is preferably between 0 and 1. -
References (PubMed ID):19642274 27074043
For DAC algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding DAC --phyche_list_file DNA/example/di_DNA_phyche_list.txt --extra_index_file DNA/example/di_DNA_indices.txt --lag 2
-
Tips:Parameter lag in DAC, lag must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 2. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):19706744 29272359
For DCC algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding DCC --phyche_list_file DNA/example/di_DNA_phyche_list.txt --extra_index_file DNA/example/di_DNA_indices.txt --lag 2
-
Tips:Parameter lag in DCC, lag must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 2. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):19706744 29272359
For DACC algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding DACC --phyche_list_file DNA/example/di_DNA_phyche_list.txt --extra_index_file DNA/example/di_DNA_indices.txt --lag 2
-
Tips:Parameter lag in DACC, lag must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 2. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):19706744 29272359
For TAC algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding TAC --phyche_list_file DNA/example/tri_DNA_phyche_list.txt --extra_index_file DNA/example/tri_DNA_indices.txt --lag 2
-
Tips:Parameter lag in TAC, lag must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 3. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):19706744 29272359
For TCC algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding TCC --phyche_list_file DNA/example/tri_DNA_phyche_list.txt --extra_index_file DNA/example/tri_DNA_indices.txt --lag 2
-
Tips:Parameter lag in TCC, lag must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 3. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):19706744 29272359
For TACC algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding TACC --phyche_list_file DNA/example/tri_DNA_phyche_list.txt --extra_index_file DNA/example/tri_DNA_indices.txt --lag 2
-
Tips:Parameter lag in TACC, lag must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 3. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):19706744 29272359
For MAC algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding MAC --phyche_list_file DNA/example/di_DNA_phyche_list.txt --lamada 2 --oli 2
-
Tips:Parameter lamada in MAC, lamada must be an integer and greater than 0 and and the range of lamada is preferably between 1 and 10. The parameter oli denotes that one kind of Oligonucleotide for DNA: 2 represents dinucleotide and 3 represents trinucleotide. The format of parameter phyche_list_file should be consistent with the corresponding sample file. -
References (PubMed ID):3359010 29272359
For GAC algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding GAC --phyche_list_file DNA/example/di_DNA_phyche_list.txt --lamada 2 --oli 2
-
Tips:Parameter lamada in GAC, lamada must be an integer and greater than 0 and and the range of lamada is preferably between 1 and 10. The parameter oli denotes that one kind of Oligonucleotide for DNA: 2 represents dinucleotide and 3 represents trinucleotide. The format of parameter phyche_list_file should be consistent with the corresponding sample file. -
References (PubMed ID):16261547 29272359
For NMBAC algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding NMBAC --phyche_list_file DNA/example/di_DNA_phyche_list.txt --lamada 2 --oli 2
-
Tips:Parameter lamada in NMBAC, lamada must be an integer and greater than 0 and and the range of lamada is preferably between 1 and 10. The parameter oli denotes that one kind of Oligonucleotide for DNA: 2 represents dinucleotide and 3 represents trinucleotide. The format of parameter phyche_list_file should be consistent with the corresponding sample file. -
References (PubMed ID):11043931 29272359
For PseDNC algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding PseDNC --lamada 2 --weight 0.1
-
Tips:Parameter lamada in PseDNC, lamada must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 2. The parameter weight must be a number and the range of weight is preferably between 0 and 1. -
References (PubMed ID):23303794 29272359
For PseKNC algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding PseKNC --lamada 2 --weight 0.1 --kValue 3
-
Tips:The parameter kValue must be an integer and the range of kValue is preferably between 1 and 6. The parameter lamada must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 3. The parameter weight must be a number and the range of weight is preferably between 0 and 1. -
References (PubMed ID):24504871 29272359
For PC-PseDNC-General algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding PC-PseDNC-General --lamada 2 --weight 0.1 --phyche_list_file DNA/example/di_DNA_phyche_list.txt --extra_index_file DNA/example/di_DNA_indices.txt
-
Tips:Parameter lamada in PC-PseDNC-General, lamada must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 2. The parameter weight must be a number and the range of weight is preferably between 0 and 1. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):24318998 29272359
For PC-PseTNC-General algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding PC-PseTNC-General --lamada 2 --weight 0.1 --phyche_list_file DNA/example/tri_DNA_phyche_list.txt --extra_index_file DNA/example/tri_DNA_indices.txt
-
Tips:Parameter lamada in PC-PseTNC-General, lamada must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 3. The parameter weight must be a number and the range of weight is preferably between 0 and 1. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):24318998 29272359
For SC-PseDNC-General algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding SC-PseDNC-General --lamada 2 --weight 0.1 --phyche_list_file DNA/example/di_DNA_phyche_list.txt --extra_index_file DNA/example/di_DNA_indices.txt
-
Tips:Parameter lamada in SC-PseDNC-General, lamada must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 2. The parameter weight must be a number and the range of weight is preferably between 0 and 1. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):24318998 29272359
For SC-PseTNC-General algorithm:
-
Command line:python calculateFeature.py --input DNA/example/example_dna.fasta --output output.csv --seqType DNA --encoding SC-PseTNC-General --lamada 2 --weight 0.1 --phyche_list_file DNA/example/tri_DNA_phyche_list.txt --extra_index_file DNA/example/tri_DNA_indices.txt
-
Tips:Parameter lamada in SC-PseTNC-General, lamada must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 3. The parameter weight must be a number and the range of weight is preferably between 0 and 1. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):24318998 29272359
RNA:
For Kmer algorithm:
-
Command line:python calculateFeature.py --input RNA/example/test_rna.fasta --output output.csv --seqType RNA --encoding Kmer --kValue 2
-
Tips:Parameter kValue in Kmer, kValue must be an integer and the range of kValue is preferably between 1 and 6. -
References (PubMed ID):26355518 29272359
For Mismatch algorithm:
-
Command line:python calculateFeature.py --input RNA/example/example_rna.fasta --output output.csv --seqType RNA --encoding Mismatch --kValue 3 --mismatch 1
-
Tips:Parameter kValue in Mismatch, kValue must be an integer and the range of kValue is preferably between 1 and 6. Parameter mismatch in Mismatch, mismatch must be an integer and greater than 0 and smaller than kValue. -
References (PubMed ID):14990442 19642274
For Subsequence algorithm:
-
Command line:python calculateFeature.py --input RNA/example/example_rna.fasta --output output.csv --seqType RNA --encoding Subsequence --kValue 3 --delta 1
-
Tips:Parameter kValue in Subsequence, kValue must be an integer and the range of kValue is preferably between 1 and 3. Parameter delta in Subsequence, delta must be a number and the range of delta is preferably between 0 and 1. -
References (PubMed ID):19642274 27074043
For DAC algorithm:
-
Command line:python calculateFeature.py --input RNA/example/example_rna.fasta --output output.csv --seqType RNA --encoding DAC --phyche_list_file RNA/example/di_RNA_phyche_list.txt --extra_index_file RNA/example/di_RNA_indices.txt --lag 2
-
Tips:Parameter lag in DAC, lag must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 2. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):19706744 29272359
For DCC algorithm:
-
Command line:python calculateFeature.py --input RNA/example/example_rna.fasta --output output.csv --seqType RNA --encoding DCC --phyche_list_file RNA/example/di_RNA_phyche_list.txt --extra_index_file RNA/example/di_RNA_indices.txt --lag 2
-
Tips:Parameter lag in DCC, lag must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 2. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):19706744 29272359
For DACC algorithm:
-
Command line:python calculateFeature.py --input RNA/example/example_rna.fasta --output output.csv --seqType RNA --encoding DACC --phyche_list_file RNA/example/di_RNA_phyche_list.txt --extra_index_file RNA/example/di_RNA_indices.txt --lag 2
-
Tips:Parameter lag in DACC, lag must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 2. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):19706744 29272359
For MAC algorithm:
-
Command line:python calculateFeature.py --input RNA/example/example_rna.fasta --output output.csv --seqType RNA --encoding MAC --phyche_list_file RNA/example/di_RNA_phyche_list_auto.txt --lamada 1
-
Tips:Parameter lamada in MAC, lamada must be an integer and greater than 0 and and the range of lamada is preferably between 1 and 10. The format of parameter phyche_list_file should be consistent with the corresponding sample file. -
References (PubMed ID):3359010 29272359
For GAC algorithm:
-
Command line:python calculateFeature.py --input RNA/example/example_rna.fasta --output output.csv --seqType RNA --encoding GAC --phyche_list_file RNA/example/di_RNA_phyche_list_auto.txt --lamada 1
-
Tips:Parameter lamada in GAC, lamada must be an integer and greater than 0 and and the range of lamada is preferably between 1 and 10. The format of parameter phyche_list_file should be consistent with the corresponding sample file. -
References (PubMed ID):16261547 29272359
For NMBAC algorithm:
-
Command line:python calculateFeature.py --input RNA/example/example_rna.fasta --output output.csv --seqType RNA --encoding NMBAC --phyche_list_file RNA/example/di_RNA_phyche_list_auto.txt --lamada 1
-
Tips:Parameter lamada in NMBAC, lamada must be an integer and greater than 0 and and the range of lamada is preferably between 1 and 10. The format of parameter phyche_list_file should be consistent with the corresponding sample file. -
References (PubMed ID):11043931 29272359
For PC-PseDNC-General algorithm:
-
Command line:python calculateFeature.py --input RNA/example/example_rna.fasta --output output.csv --seqType RNA --encoding PC-PseDNC-General --lamada 2 --weight 0.1 --phyche_list_file RNA/example/di_RNA_phyche_list.txt --extra_index_file RNA/example/di_RNA_indices.txt
-
Tips:Parameter lamada in PC-PseDNC-General, lamada must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 2. The parameter weight must be a number and the range of weight is preferably between 0 and 1. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):25231908 29272359
For SC-PseDNC-General algorithm:
-
Command line:python calculateFeature.py --input RNA/example/example_rna.fasta --output output.csv --seqType RNA --encoding SC-PseDNC-General --lamada 2 --weight 0.1 --phyche_list_file RNA/example/di_RNA_phyche_list.txt --extra_index_file RNA/example/di_RNA_indices.txt
-
Tips:Parameter lamada in SC-PseDNC-General, lamada must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 2. The parameter weight must be a number and the range of weight is preferably between 0 and 1. The formats of parameter phyche_list_file and extra_index_file should be consistent with the corresponding sample file. -
References (PubMed ID):25231908 29272359
For Triplet algorithm:
-
Command line:python calculateFeature.py --input RNA/example/example_rna.sc --output output.csv --seqType RNA --encoding Triplet
-
Tips:.sc file is generated by ViennaRNA-2.4.8. -
References (PubMed ID):16381612 29272359
For PseSSC algorithm:
-
Command line:python calculateFeature.py --input RNA/example/example_rna.sc --output output.csv --seqType RNA --encoding PseSSC --kValue 1 --lamada 1 --weight 0.5
-
Tips:.sc file is generated by ViennaRNA-2.4.8. The parameter kValue must be an integer and the range of kValue is preferably between 1 and 6. The parameter lamada must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 3. The parameter weight must be a number and the range of weight is preferably between 0 and 1. -
References (PubMed ID):25821974 29272359
For PseDPC algorithm:
-
Command line:python calculateFeature.py --input RNA/example/example_rna.sc --output output.csv --seqType RNA --encoding PseDPC --kValue 1 --lamada 1 --weight 0.5
-
Tips:.sc file is generated by ViennaRNA-2.4.8. The parameter kValue must be an integer and greater than 0 or equal to 0 and smaller than that minimum length of submitted sequences minus 1. The parameter lamada must be an integer and greater than 0 and smaller than that minimum length of submitted sequences minus 1. The parameter weight must be a number and the range of weight is preferably between 0 and 1. -
References (PubMed ID):25645238 29272359
2.4 Intermediate file generation
For protein descriptors, there are four types intermediate file are required, including .pssm file, .dis file, .ss2 file and .spXout file. For PSSM-based features, the corresponding algorithm can traverse sequence itself to make sequence and .pssm file one to one. But other features that require intermediate files, the name of sequence must be consistent with the name of the corresponding intermediate file. Facing the stringent format problems, we provide the user with three python scripts in util folder to generate formatted three intermediate files (.dis file, .ss2 file and .spXout file). The executed command line as follows:
python generateDisorder.py --file ../Protein/example/example_peptide.fasta --vsl2 /var/www/cgi-bin/VSL2/VSL2.jar --out ../Protein/example/disorder_files/ python generateSecondaryStructure.py --file ../Protein/example/example_peptide.fasta --psipred /var/www/cgi-bin/psipred/runpsipred --out ../Protein/example/ss_files/ python generateSpineX.py --file ../Protein/example/example_peptide.fasta --spineX /var/www/cgi-bin/spineXpublic/spX.pl --out ../Protein/example/spX_files/
2.5 Annotation of the computational results:
-
computational results are represented in
csvformat.